Ricercatori hanno scoperto un meccanismo che protegge le cellule nervose dalla morte prematura (ferroptosi), dandoci la prima prova molecolare che la ferroptosi può guidare la neurodegenerazione nel cervello umano. Questi risultati aprono nuove strade per lo sviluppo di terapie future, in particolare per la demenza infantile grave a esordio precoce.
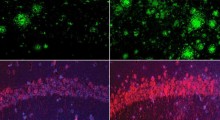
 e dei nuclei cellulari (blu). Fonte: Helmholtz Munich") Cellule cerebrali coltivate in laboratorio derivate da cellule staminali umane: i colori evidenziano le caratteristiche tipiche dei neuroni corticali (verde e rosso) e dei nuclei cellulari (blu). Fonte: Helmholtz Munich
Cellule cerebrali coltivate in laboratorio derivate da cellule staminali umane: i colori evidenziano le caratteristiche tipiche dei neuroni corticali (verde e rosso) e dei nuclei cellulari (blu). Fonte: Helmholtz Munich
Perché i neuroni muoiono nella demenza, ed è possibile rallentare questo processo? Un team internazionale guidato dal Prof. Marcus Conrad dell’Helmholtz Monaco (Germania), titolare della cattedra di Biologia Redox Traslazionale all’Università Tecnica di Monaco (TUM), descrive ora in Cell come i neuroni si proteggono dalla morte ferroptotica delle cellule.
Centrale in questo meccanismo di difesa è il selenoenzima 'glutatione perossidasi 4' (GPX4). Una singola mutazione del gene che codifica GPX4, può interrompere una componente cruciale e finora sconosciuta della funzione dell’enzima. Nei bambini colpiti, ciò porta a una grave demenza ad esordio precoce. Quando è pienamente funzionale, GPX4 inserisce un breve anello proteico – una sorta di 'pinna' – nella parte interna della membrana cellulare neuronale, consentendo all’enzima di neutralizzare le sostanze nocive note come perossidi lipidici.
Navigare lungo la membrana cellulare
"GPX4 è un po' come una tavola da surf", afferma Conrad. "Con la sua pinna immersa nella membrana cellulare, scorre lungo la superficie interna e disintossica rapidamente i perossidi lipidici mentre avanza".
Una singola mutazione puntiforme trovata nei bambini con demenza a esordio precoce altera questo anello proteico a forma di pinna: l’enzima non può più inserirsi correttamente nella membrana per svolgere la sua funzione protettiva. I perossidi lipidici sono quindi liberi di danneggiare la membrana, innescando la ferroptosi e la rottura cellulare, e i neuroni muoiono.
Lo studio è iniziato con tre bambini negli Stati Uniti che soffrono di una forma estremamente rara di demenza infantile precoce. Tutti e tre portano lo stesso cambiamento nel gene GPX4, la mutazione R152H. Lavorando su campioni di cellule di un bambino affetto, i ricercatori sono riusciti a studiare gli effetti della mutazione in modo più dettagliato e di riprogrammare le cellule in uno stato simile a quello delle cellule staminali. Da queste cellule staminali riprogrammate, hanno poi generato neuroni corticali e strutture tissutali tridimensionali che somigliano al tessuto cerebrale iniziale, i cosiddetti organoidi cerebrali.
Le prove di laboratorio confermano: senza GPX4 funzionale, si sviluppa la demenza
Per capire cosa succede a livello dell’intero organismo, il team ha poi introdotto la mutazione R152H in topi modello, alterando così specificamente l’enzima GPX4 in diversi tipi di cellule nervose. Come risultato della compromissione della funzione GPX4, gli animali hanno gradualmente sviluppato gravi deficit motori, con neuroni morenti nella corteccia cerebrale e nel cervelletto e risposte neuroinfiammatorie pronunciate nel cervello, un modello che rispecchia da vicino le osservazioni nei bambini affetti e ricorda fortemente i profili delle malattie neurodegenerative.
Parallelamente, i ricercatori hanno analizzato quali proteine cambiano in abbondanza nel modello sperimentale. Hanno osservato uno schema sorprendentemente simile a quello osservato nei pazienti con morbo di Alzheimer (MA): numerose proteine che aumentano o calano nel MA erano similmente disregolate nei topi privi di GPX4 funzionale. Ciò suggerisce che lo stress ferroptotico può avere un ruolo non solo in questo raro disturbo ad esordio precoce, ma potenzialmente anche in forme più comuni di demenza.
Una nuova visione sulle cause della demenza
"I nostri dati indicano che la ferroptosi può essere una forza trainante dietro la morte neuronale, non solo un effetto collaterale", afferma la dott.ssa Svenja Lorenz, una dei primi autori dello studio. "Fino ad ora, la ricerca sulla demenza si è spesso concentrata sui depositi di proteine nel cervello, le cosiddette placche di amiloide-β. Ora stiamo ponendo di più l'accento sul danno alle membrane cellulari che mette in moto questa degenerazione".
Gli esperimenti iniziali mostrano anche che la morte cellulare innescata dalla perdita di GPX4 può essere rallentata in colture cellulari e nei topi modello usando composti che inibiscono specificamente la ferroptosi. Il dottor Tobias Seibt, nefrologo all'Ospedale universitario LMU di Monaco e primo coautore, afferma: "Questa è un'importante prova di principio, ma non è ancora una terapia".
Il dottor Adam Wahida, anch'egli uno dei primi autori dello studio, aggiunge: "A lungo termine, possiamo immaginare strategie genetiche o molecolari per stabilizzare questo sistema protettivo. Per ora, tuttavia, il nostro lavoro rimane chiaramente nell'ambito della ricerca di base".
Fonte: Helmholtz Munich (> English) - Traduzione di Franco Pellizzari.
Riferimenti: SM Lorenz, [+71], M Conrad. A fin-loop-like structure in GPX4 underlies neuroprotection from ferroptosis. Cell, 2025, DOI
Copyright: Tutti i diritti di testi o marchi inclusi nell'articolo sono riservati ai rispettivi proprietari.
Liberatoria: Questo articolo non propone terapie o diete; per qualsiasi modifica della propria cura o regime alimentare si consiglia di rivolgersi a un medico o dietologo. Il contenuto non rappresenta necessariamente l'opinione dell'Associazione Alzheimer OdV di Riese Pio X ma solo quella dell'autore citato come "Fonte". I siti terzi raggiungibili da eventuali collegamenti contenuti nell'articolo e/o dagli annunci pubblicitari sono completamente estranei all'Associazione, il loro accesso e uso è a discrezione dell'utente. Liberatoria completa qui.
Nota: L'articolo potrebbe riferire risultati di ricerche mediche, psicologiche, scientifiche o sportive che riflettono lo stato delle conoscenze raggiunte fino alla data della loro pubblicazione.