I risultati puntano a un farmaco promettente che ripristina l’equilibrio neuronale e può rallentare la progressione della malattia
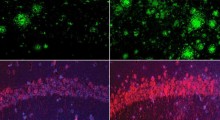
") A sinistra neuroni sani, a destra neuroni con gene KCNQ2 con giuntura errata (Fonte: Joseph et al / NatureNeurosci)
A sinistra neuroni sani, a destra neuroni con gene KCNQ2 con giuntura errata (Fonte: Joseph et al / NatureNeurosci)
Un nuovo studio della Northwestern University di Evanston (Illinois/USA) su tessuto nervoso di pazienti e su neuroni umani coltivati in laboratorio, ha scoperto che una proteina chiave di malattia, la TDP-43, guida l'iperattività delle cellule nervose nelle malattie neurodegenerative sclerosi laterale amiotrofica (SLA) e demenza frontotemporale (FTD).
I risultati non solo spiegano un mistero di vecchia data sul perché le cellule nervose si attivano eccessivamente nella SLA e nella FTD, ma evidenziano anche un nuovo farmaco promettente per rallentare o prevenire la progressione della malattia.
La SLA, che attacca i motoneuroni del midollo spinale e provoca debolezza progressiva e atrofia muscolare, colpisce circa 350.000 persone in tutto il mondo. La FTD porta all’atrofia dei lobi frontali e temporali del cervello, che sono noti per controllare la personalità, il comportamento e il linguaggio. Si stima che da 1,2 a 1,8 milioni di persone in tutto il mondo convivano con una qualche forma di FTD.
Sebbene la SLA e la FTD siano piuttosto diverse, una caratteristica coerente ma poco spiegata di entrambe le malattie è l’ipereccitabilità neuronale, in cui i neuroni si attivano troppo e troppo facilmente. Precedenti ricerche avevano scoperto che quasi tutti i casi di SLA e circa la metà dei casi di FTD condividono un problema caratteristico: la proteina TDP-43 si sposta dalla sua posizione normale (il nucleo) al citoplasma, e interrompe la normale funzione cellulare.
Il nuovo studio ha scoperto che quando la TDP-43 non funziona correttamente, interrompe il normale splicing (giuntura) del canale KCNQ2, che può essere considerato un 'freno' che in genere impedisce ai neuroni di attivarsi troppo. Senza questo freno, i neuroni diventano elettricamente iperattivi (ipereccitabilità neuronale). È stato dimostrato che i pazienti affetti da SLA che presentano ipereccitabilità hanno un aumento del rischio di mortalità, sottolineandone il significato clinico.
Gli autori dello studio hanno progettato, sviluppato e testato un farmaco punta-geni che, secondo lo studio, può correggere questo errore nei neuroni umani coltivati in laboratorio, ripristinando l’equilibrio e riducendo l’iperattività. Il farmaco, chiamato oligonucleotide antisenso (ASO), una volta validato e approvato clinicamente, verrebbe somministrato tramite iniezione diretta nel sistema nervoso centrale del paziente, come una epidurale.
"Correggendo l'errore di splicing del KCNQ2 con il farmaco ASO, abbiamo calmato i neuroni iperattivi e il ripristino dell'attività neuronale potrebbe potenzialmente rallentare la progressione della malattia", ha affermato l'autore corrispondente Evangelos Kiskinis, professore associato di neuroscienze e neurologia nella divisione di malattie neuromuscolari della Northwestern University. “Sono entusiasta che abbiamo finalmente risolto un mistero di lunga data sul perché le cellule nervose nella SLA/FTD sono iperattive e stressate anche prima di morire”.
Gli scienziati hanno studiato neuroni di pazienti e altri coltivati in laboratorio, nonché il tessuto cerebrale e del midollo spinale post-mortem della SLA e della FTD. Il difetto che hanno scoperto è specifico degli esseri umani e non si verifica nei modelli di topo o ratto, ha detto Kiskinis. I pazienti nello studio con splicing errato del KCNQ2 più grave avevano un esordio precoce della malattia, ha scoperto lo studio, rendendolo un possibile biomarcatore per la prognosi o la risposta al trattamento, ha detto Kiskinis.
“Il nostro lavoro collega due caratteristiche centrali della malattia – la patologia TDP-43 e l’ipereccitabilità – in un unico percorso meccanicistico”, ha detto Kiskinis. “Indica anche un nuovo entusiasmante obiettivo terapeutico”.
Il team di Kiskinis sta attualmente lavorando per sviluppare un test sui biomarcatori basato sull’identificazione di questo evento KCNQ2 a giuntura errata che potrebbe portare a una diagnosi più precoce. “Siamo entusiasti di portare questo ASO nelle fasi cliniche”, ha affermato Kiskinis.
Fonte: Kristin Samuelson in Northwestern University (> English) - Traduzione di Franco Pellizzari.
Riferimenti: BJ Joseph, [+35], E Kiskinis. TDP-43-dependent mis-splicing of KCNQ2 triggers intrinsic neuronal hyperexcitability in ALS/FTD. Nat Neurosci, 2025, DOI
Copyright: Tutti i diritti di testi o marchi inclusi nell'articolo sono riservati ai rispettivi proprietari.
Liberatoria: Questo articolo non propone terapie o diete; per qualsiasi modifica della propria cura o regime alimentare si consiglia di rivolgersi a un medico o dietologo. Il contenuto non rappresenta necessariamente l'opinione dell'Associazione Alzheimer OdV di Riese Pio X ma solo quella dell'autore citato come "Fonte". I siti terzi raggiungibili da eventuali collegamenti contenuti nell'articolo e/o dagli annunci pubblicitari sono completamente estranei all'Associazione, il loro accesso e uso è a discrezione dell'utente. Liberatoria completa qui.
Nota: L'articolo potrebbe riferire risultati di ricerche mediche, psicologiche, scientifiche o sportive che riflettono lo stato delle conoscenze raggiunte fino alla data della loro pubblicazione.