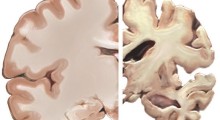
SIRT2, enzima finora sconosciuto nella produzione di GABA degli astrociti, può contenere la chiave per separare gli effetti delle molecole degenerative nell'Alzheimer.
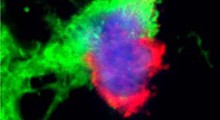
. SIRT2 è l'ultimo enzima nel percorso di produzione GABA (a destra). L'inibizione di SIRT2 riduce GABA, evitando l'inibizione neuronale e la perdita di memoria, ma la produzione di H2O2 continua, contribuendo alla morte dei neuroni. (Fonte: Bhalla_et_al / Molecular Neurodegeneration)") Gli astrociti reattivi nell'Alzheimer assorbono e abbattono le placche amiloidi con l'autofagia, provocando sovraregolazione del ciclo dell'urea e produzione di putrescina-a-Gaba. Ciò porta a una produzione elevata di GABA e H2O2 tossico, che inibisce la segnalazione neuronale e provoca la morte neuronale (a sinistra). SIRT2 è l'ultimo enzima nel percorso di produzione GABA (a destra). L'inibizione di SIRT2 riduce GABA, evitando l'inibizione neuronale e la perdita di memoria, ma la produzione di H2O2 continua, contribuendo alla morte dei neuroni. (Fonte: Bhalla_et_al / Molecular Neurodegeneration)
Gli astrociti reattivi nell'Alzheimer assorbono e abbattono le placche amiloidi con l'autofagia, provocando sovraregolazione del ciclo dell'urea e produzione di putrescina-a-Gaba. Ciò porta a una produzione elevata di GABA e H2O2 tossico, che inibisce la segnalazione neuronale e provoca la morte neuronale (a sinistra). SIRT2 è l'ultimo enzima nel percorso di produzione GABA (a destra). L'inibizione di SIRT2 riduce GABA, evitando l'inibizione neuronale e la perdita di memoria, ma la produzione di H2O2 continua, contribuendo alla morte dei neuroni. (Fonte: Bhalla_et_al / Molecular Neurodegeneration)
Un team di ricerca dell'Institute for Basic Science (IBS) di Daejeon, Corea del Sud, ha identificato un enzima finora sconosciuto, SIRT2, che ha un ruolo chiave nella perdita di memoria associata al morbo di Alzheimer (MA). Lo studio, pubblicato su Molecular Neurodegeneration e guidato da C. Justin Lee, direttore del Center for Cognition and Sociality, fornisce informazioni critiche su come gli astrociti contribuiscono al declino cognitivo, producendo quantità eccessive del neurotrasmettitore inibitorio GABA.
Gli astrociti, che un tempo si pensava avessero solo un ruolo di supporto ai neuroni, sono ora noti per influenzare attivamente la funzione cerebrale. Nel MA, gli astrociti diventano reattivi, il che significa che cambiano il comportamento in risposta alla presenza di placche amiloide-beta (Aβ), un segno distintivo della malattia. Mentre gli astrociti tentano di eliminare queste placche, il processo innesca una reazione a catena dannosa.
Prima, come scoperto da ricerche precedenti, assorbono le placche tramite autofagia (rif.3) e le degradano dal ciclo dell'urea (rif 2). Ma questa scomposizione si traduce nella sovrapproduzione di GABA, che smorza l'attività cerebrale e porta a compromissione della memoria. Inoltre, questo percorso genera perossido di idrogeno (H2O2), un sottoprodotto tossico che provoca ulteriore morte neuronale e neurodegenerazione.
Il team di ricerca dell'IBS ha deciso di scoprire quali enzimi erano responsabili dell'eccessiva produzione di GABA, sperando di trovare un modo per bloccare selettivamente i suoi effetti dannosi senza interferire con altre funzioni cerebrali. Usando analisi molecolari, scansioni al microscopio ed elettrofisiologia, i ricercatori hanno identificato SIRT2 e ALDH1A1 come enzimi critici coinvolti nella sovrapproduzione di GABA negli astrociti colpiti dal MA.
Si è scoperto che la proteina SIRT2 è aumentata negli astrociti dei topi modello di MA usati di solito e nel cervello dei pazienti di MA post mortem.
"Quando abbiamo inibito l'espressione astrocitica di SIRT2 nei topi con MA, abbiamo osservato il recupero parziale della memoria e la riduzione della produzione di GABA", ha scritto Mridula Bhalla, prima autrice dello studio e ricercatrice post-dottorato dell'IBS. "Sebbene ci aspettassimo un rilascio ridotto di GABA, abbiamo scoperto che era stata recuperata solo la memoria di lavoro a breve termine (labirinto Y) dei topi, mentre la memoria spaziale (NPR) non lo era. Questo è stato interessante ma ci ha anche lasciato più domande".
Il SIRT2 partecipa all'ultima fase della produzione di GABA, mentre l'H2O2 viene prodotto in precedenza nel processo. È quindi possibile che l'H2O2 sia prodotto e rilasciato costantemente dalle cellule anche in assenza di SIRT2.
"In effetti, abbiamo scoperto che l'inibizione del SIRT2 ha continuato la produzione di H2O2, indicando che la degenerazione neuronale potrebbe continuare anche se la produzione di GABA si riduce", afferma il direttore C. Justin Lee.
Identificando SIRT2 e ALDH1A1 come obiettivi a valle, gli scienziati possono ora inibire selettivamente la produzione di GABA senza influire sui livelli di H2O2. Questa è una svolta cruciale perché consente ai ricercatori di separare gli effetti di GABA e H2O2 e di studiare i loro ruoli individuali nella neurodegenerazione. Il direttore C. Justin Lee ha sottolineato l'importanza di questi risultati, affermando:
“Finora, nella ricerca del MA abbiamo usato gli inibitori dell'enzima monoamine oxidase B (MAOB), che bloccano la produzione di H2O2 e GABA. Identificando gli enzimi SIRT2 e ALDH1A1 a valle del MAOB, ora possiamo inibire selettivamente la produzione di GABA, che ci permetterebbe di sezionare gli effetti di GABA e H2HO e studiare i loro ruolli individuali nella progressione della malattia".
Mentre SIRT2 potrebbe non essere un obiettivo diretto di farmaci a causa dei suoi effetti limitati sulla neurodegenerazione, questa ricerca apre la strada a strategie terapeutiche più precise volte a controllare la reattività astrocitica nel MA.
Fonte: IBS-Institute for Basic Science (> English) - Traduzione di Franco Pellizzari.
Riferimenti:
- M Bhalla, [+11], CJ Lee. SIRT2 and ALDH1A1 as critical enzymes for astrocytic GABA production in Alzheimer’s disease. Molecular Neurodeg, 2025, DOI
- Y Ju, et al. Astrocytic urea cycle detoxifies Aβ-derived ammonia while impairing memory in Alzheimer’s disease. Cell Metabolism, 2022, DOI
- S Kim, et al. Astrocytic autophagy plasticity modulates Aβ clearance and cognitive function in Alzheimer’s disease. Molecular Neurodeg, 2024, DOI
Copyright: Tutti i diritti di testi o marchi inclusi nell'articolo sono riservati ai rispettivi proprietari.
Liberatoria: Questo articolo non propone terapie o diete; per qualsiasi modifica della propria cura o regime alimentare si consiglia di rivolgersi a un medico o dietologo. Il contenuto non rappresenta necessariamente l'opinione dell'Associazione Alzheimer OdV di Riese Pio X ma solo quella dell'autore citato come "Fonte". I siti terzi raggiungibili da eventuali collegamenti contenuti nell'articolo e/o dagli annunci pubblicitari sono completamente estranei all'Associazione, il loro accesso e uso è a discrezione dell'utente. Liberatoria completa qui.
Nota: L'articolo potrebbe riferire risultati di ricerche mediche, psicologiche, scientifiche o sportive che riflettono lo stato delle conoscenze raggiunte fino alla data della loro pubblicazione.